前言
在后面的章节中,我们将介绍基因组学实验中的统计推断。我们会使用前面几节中介绍的一些概念,其中包括t检验和多重比较。在这一节中,我们先介绍一个在基因组学数据分析中一个极为重要的概念:生物学变异与技术变异(biological and technical variability)以及它们之间的区别。
通常而言,我们在一个种群里所观察到的生物学单位,例如某个个体的变异称为生物学变异(biological variability)。我们将那些对同一个生物学单位进行测量时所观察到的变异称为技术性变异(technical variability,例如同一个样本测3次,这3次就是技术重复,3次数据的变异称为技术变异)。由于在基因组学研究中经常会用到新技术,因此技术重复(technical replicates)在很多时候都会用于评估实验数据。理想情况下,同一样本的检测结果应该是相同的,但在实际检测中同一样本的多次检测会出现一定的偏差,因此使用技术重复我们就能我们就能够评估新技术的技术变异。我还经常使用生物学重复(biological replicates)与技术重复(technical replicates)来分别代指生物学变异和技术变异。
我们在进行统计推断来解释有差异时,需要注意不要混淆生物学变异和技术变异,因为它们所代表的意义是不同的。例如,当我们分析技术重复时,样本仅仅只有一个,而不是代表某个总体,例如人类或小鼠。这一节中,我们通过设计一个实验来说明一下技术重复和生物重复。
合并实验数据
我们将要研究的数据集来源于基因表达微阵列。在这个实验中,我们从两个品系的小鼠中分别随机选择了12只小鼠,并提取了它们的RNA。我们将得到的24个样本使用微阵列的手段进行检测,引外,我们还构成了两个库,这两个库中的样本则是分别来源于这两个品系的12只小鼠的混合样本。
现在我们安装以下包,这个包中含有这些数据,如下所示:
|
|
使用pData函数我们就能查看实验设计。每行表示一个样本,每列表示1只小鼠。例如i,j单元格,中如果是1,则表示RNA数据来源于j只小鼠的样本i。通过行名我们可以知道品系信息(不推荐这种方法来进行分组,你可以向phenoData中添加一个变量,用于指定小鼠品系信息),数据如下所示:
|
|
现在我们创建一个函数,用于说明小鼠的样本信息,如下所示:
|
|
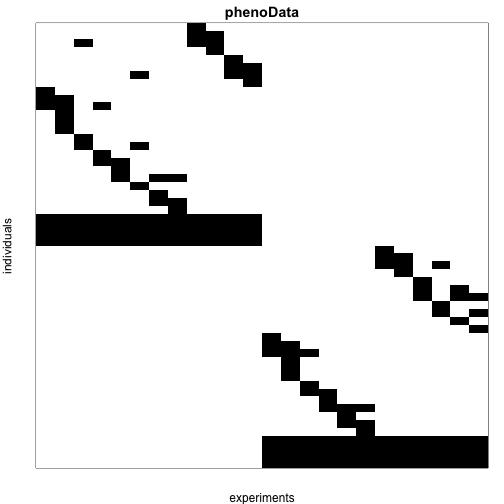
需要注意的是,最终我们只对两个品系小鼠的差异基因感兴趣,也就是数据中标为0的品系和1的品系。我们可以对合并样本的技术重复进行检验或来源于12只小鼠的数据进行检验。我们可以提取出这些合并的样本,因为来自于每个品系的所有小鼠都都可以用0或1来表示,因此那些来源于某品系(例如用1表示的这个品系)代表了这些样本,因此我们提取出矩阵行之和为12的基因,这段话不太理解,原文如下:
Note that ultimately we are interested in detecting genes that are differentially expressed between the two strains of mice which we will refer to as strain 0 and 1. We can apply tests to the technical replicates of pooled samples or the data from 12 individual mice. We can identify these pooled samples because all mice from each strain were represented in these samples and thus the sum of the rows of experimental design matrix add up to 12:
如下所示:
|
|
我们可以从列名确定品系,如下所示:
|
|
如果我们要比较每个基因在不同品系之间的差异,我们就会发现有几个基因出现了比较一致的差异,如下所示:
|
|
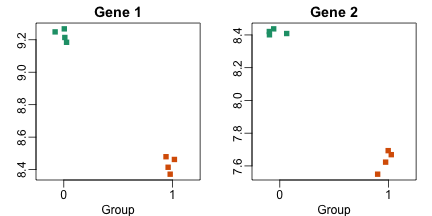
这里要注意一下,如果我们根据这些值来进行t检验,那么我们就会得到非常显著的结果,也就是说p值极低,如下所示:
|
|
但是,如果我们再次从两个品系的小鼠中各选择12个小鼠的话,这样的结论会成立吗?请注意,t检验的定义包括要比较的总体的标准差。我们这里做的计算结果是否能够反映出总体差异?(注:这里要回顾一下t检验的定义,样本与方差的差异等概念)。
需要注意的是,我们现在重复计算的过程就是在重复原来的实验流程。我们为每个合并的样本创建了4个技术重复。或许Gene 1在小鼠品系内存在着很高的变异,而Gene 2的变异则比较低,但是我们不能看到这一点,因为小鼠之间的变异被合并后的数据所掩盖了(注:这里作者只是假设了Gene 1可能存在着比较高的组内变异)。
我们还有每只小鼠的微阵列数据。对于每个品系的小鼠来说,我们有12个生物学重复。我们可以通过查询1来找到这个品系,如下所示:
|
|
事实证明,某些单独的小鼠本身就含有技术重复,因此现在我们将这种情况剔除掉,从而用于说明仅用生物学重复的分析结果,如下所示:
|
|
我们可以计算每个基因的在某个品系中的方差,并与通过技术重复获得的标准差进行比较,如下所示:
|
|
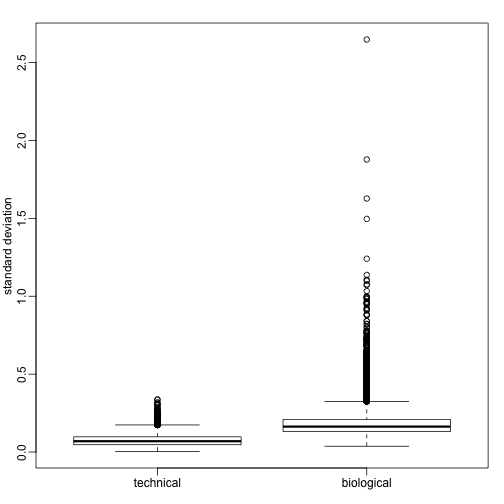
从上图上我们可以看到,生物学方差(variance)远大于技术方差。并且对于方差的变异而言,生物学方差更大。这只是我们前面提到的2个基因的情况,现在我们展示每只小鼠上测得的这2个基因的表达值,如下所示:
|
|
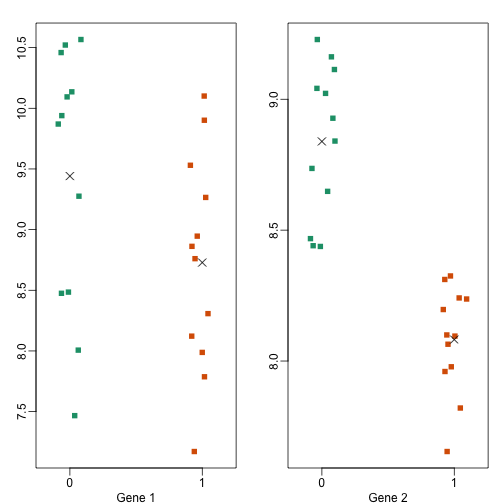
现在p值就发生了一点变化,如下所示:
|
|
当我们在不同品系的小鼠之间比较这2个基因的差异时,我们对哪个基因的差异更有信心呢(Gene 1还是Gene 2)?如果另一位研究人员另外一批随机样本来做实验,你认识他会发现哪个基因有差异(Gene 1还是Gene 2)?如果我们希望我们的结论是关于小鼠品系的通用结论(通用结论这里我的理解就是,无论放哪个实验室做,都能做出这个结果,得到这个结论),而不仅仅是我们这次实验的结论(也就是说,有可能就是只有自己实验室能做出这个结果,那么这个结果就不太可信,有可能是假阳性),那么检测生物学的变异则非常重要。
在这一节中,我们使用了两种分析方法。第一种分析方法是将这两个品系的生物学重复做为总体总体来进行分析析(也就是说把某个品系的12只小鼠数据汇总起来分析)。第二种则是将合并后的数据来作为总体进行分析(我的理解就是把12只小鼠的RNA混合在一起,作为一个样本,然后使用4个技术重复来进行分析,这种分析方法其实检测的就是技术变异)。在科学研究中,我们通常关注的是总体(也就是第一种分析方法)。作为一个非常实际的例子,这里需要注意的是,如果另外一个实验室重复该实验,他们会有另外一组12个小鼠的样本,因此关于我们实验中对总体的推断也会在另外的实验室中得到重复。
An analysis with biological replicates has as a population these two strains of mice. An analysis with technical replicates has as a population the twelve mice we selected and the variability is related to
the measurement technology. In science we typically are concerned with populations. As a very practical example, note that if another lab performs this experiment they will have another set of twelve mice and thus inferences about populations are more likely to be reproducible.